Experiment 11: Infrarot- und Raman-Spektroskopie#
Hintergrund#
Infrarot- und Raman-Spektroskopie sind Formen der Schwingungsspektroskopie und basieren auf Übergängen zwischen den quantisierten Schwingungszuständen eines Moleküls. Mit quantenchemischen Methoden können sowohl die Schwingungsfrequenzen der verschiedenen Schwingungsmoden, als auch deren Übergangswahrscheinlichkeiten (Intensitäten) bei IR- und Raman-Spektroskopie berechnet werden. Ziel dieses Experiment ist die Vorhersage von Schwingungspektren bei zunehmend komplexeren Molekülen.
Schwingungsfrequenzen#
Bei Molekülschwingungen handelt es sich um eine Bewegungen der Atomkerne relativ zu einander. Hier kann erneut die (sehr gute) Born-Oppenheimer Näherung (Elektronen folgen den Kernen instantan) getroffen werde, um die Bewegung von Kernen und Elektronen zu separieren. Die Potentialhyperfläche \(E(\vec{R})\) ist dann ausschließlich von den Kernkoordinaten \(\vec{R}\) abhängig und kann, wie auch schon für die Geometrieoptimierung, als Taylorreihe um die aktuelle Geometrie \(\vec{R}_{0}\) entwickelt werden (siehe Gleichung (2) in Suche nach Minima). Die Reihenentwicklung wird für gewöhnlich nach der zweiten Ordnung abgebrochen. Zusätzlich der Term erster Ordnung fällt weg, wenn die Geometrie \(\vec{R}_{0}\) vor der Frequenzberechnung optimiert wurde (Gradient \(\vec{g}(R_{0})=0\)). In diesem Modell verhält sich die Energie
bei der Auslenkung von \(\vec{R}_{0}\) nach \(\vec{R}\) entlang einer Schwingungsmode dem harmonischen Oszillator entsprechend quadratisch (mit den höheren Termen kann auch die Anharmonizität der Schwingungen berücksichtigt werden). Der direkte Vergleich mit dem harmonischen Oszillator
zeigt die Verbindung zwischen den harmonischen Schwingungsfrequenzen ω und der Hessematrix \(\mathbf{H}\). Explizit sind die Eigenwerte \(\varepsilon_{i}\) der massengewichteten Hessematrix
mit den atomaren Massen \(M_{j}\) und den Einträgen der Hessematrix \(H_{ij}\) proportional zum Quadrat der harmonischen Frequenzen \(\nu_{i}\). Die harmonischen Frequenzen \(\nu_{i}\) sind im Allgemeinen reell, können aber auch imaginär werden, wenn der gefundene stationäre Punkt bei der Geometrieoptimierung ein Sattelpunkt und kein Minimum ist (oder die Optimierung nicht vollständig konvergiert ist). Die Eigenvektoren von \(F_{ij}\) entsprechen den massengewichteten Normalkoordinaten \(\vec{q}\) entlang derer die verschiedenen Schwingungen verlaufen. Die Normalmoden sind folglich Linearkombinationen aus allen möglichen Schwingungen einzelner Atompaare und beinhalten das gesamte Molekül.
Für Standardmethoden wie Hartree-Fock und DFT ist die Hessematrix \(\mathbf{H}\) als geschlossener analytischer Ausdruck implementiert, was deren (immer noch teure) Berechnung beschleunigt. Abgesehen von einer analytischen Implementierung kann die Hessematrix aber auch numerisch durch Ableitung des Gradient \(\vec{g}\) bestimmt werden:
Intensitäten#
Neben den Schwingungsfrequenzen sind auch die Intensitäten notwendig, um ein IR- oder Ramam-Spektrum aufzutragen. Bei der IR-Spektroskopie wird das Molekül durch Absorption eines Photons im infraroten Bereich des Spektrums aus dem Schwingungsgrundzustand angeregt. Dabei koppelt das elektromagnetische Feld an die Änderung des Dipolmoments \(\vec{\mu}\) (erste Ableitung der Energie nach dem elektrischen Feld \(\vec{E}\)) entland der Normalkoordinate \(\vec{q}\). Es gilt unter Annahme der harmonischen Näherung:
Die Raman-Spektroskopie hingegen basiert auf der inelastischen Streuung eines Photons aus dem optischen Bereich des Spektrums (monochromatische Laserstrahlung) am Molekül und dem daraus folgenden spektralen Shift der Streustrahlung. Dabei ist für die inelastische Streuung eine Änderung der Polarisierbarkeit \(\vec{\alpha}\) (zweite Ableitung der Energie nach dem elektrischen Feld) des Moleküls entlang der Normalmode erforderlich. In harmonischer Näherung gilt dann
Durch die Abhängigkeit von der Änderung des Dipolmoments für IR- und der Polarisierbarkeit für Raman-Spektroskopie ergeben sich entgegengesetzte Auswahlregeln. Welche Schwingungen IR- und welche Raman-aktiv sind, lässt sich dabei anhand von gruppentheoretischen Überlegungen bestimmen.
Beschreibung des Experiments#
1. Schwingungsspektroskopie an kleinen Molekülen:
Zunächst sollen die Schwingungsspektren der folgenden zweiatomigen Moleküle berechnet werden: CO, HF, ClF, N2, HCl und Cl2. Als Referenz sind hier genaue experimentelle Referenzmessungen in der Gasphase vorhanden. Das Testen von theoretischen Methoden gegen experimentelle (oder theoretische) Referenzwerte hoher Genauigkeit wird als „Benchmarking“ bezeichnet und ist ein essentieller Schritt bei der Auswahl einer geeigneten Methode für eine theoretische Studie.
Molekül |
\(\tilde{\nu}_{\mathrm{exp}}/\)cm-1 |
|---|---|
CO |
2170 |
HF |
4138 |
ClF |
786 |
N2 |
2359 |
HCl |
2991 |
Cl2 |
560 |
Die Frequenzen sollen mit der composite-Methode r2-SCAN-3c berechnet werden. Diese nutzt das meta-GGA Dichtefunktional r2-SCAN mit der D4 Dispersionskorrektur und der geometrischen Counter-Poise Korrektur in der modifizierten triple-ζ Basissatz def2-mTZVPP. Folgen Sie zur Berechnung der Schwingungsspektren mit r2-SCAN-3c den folgenden Schritten:
Damit die harmonisch Näherung angenommen werden kann, muss zunächst das Minimum der Potentialhyperfläche (PES) gefunden werden. Um eine möglichst glatte PES zu erhalten, verwenden Sie das große numerische Integrationsgrid
DEFGRID3und sehr enge Konvergenzkriterien für das SCF (VeryTightSCF). Gleiches gilt auch für die Konvergenz der Geometrieoptimierung (VeryTightOpt), um imaginäre Moden bei der Frequenzrechnung zu vermeiden. Optimieren Sie nun die Geometrien mit r2-SCAN-3c und den angegebenen Einstellungen:
! R2SCAN-3C VeryTightSCF VeryTightOpt DefGrid3
%pal
nprocs 4
end
* xyzfile 0 1 struc.xyz
Bei der Berechnung von Schwingungsfrequenzen in ORCA werden IR-Intensitäten automatisch bestimmt. Für Raman-Aktivitäten werden hingegen die Polarisierbarkeit α entlang der Normalmoden benötigt, die in ORCA explizit angeforder werden muss (
% elprop Polar true end). Dies ist in ORCA nicht möglich mit einer analytischen Hessematrix und erfordert eine numerische Frequenzrechnung (NumFreq). Berechnen Sie nun für die zuvor optimierten Geometrien die numerischen Schwingungsfrequenzen und IR- sowie Raman-Intensitäten mit r2-SCAN-3c in den selben Einstellungen wie zuvor:
! R2SCAN-3C VeryTightSCF DefGrid3
! NumFreq
% elprop
Polar true
end
%pal
nprocs 4
end
* xyzfile 0 1 struc.xyz
Vergleichen Sie zunächst die harmonisch berechneten Frequenzen mit den experimentell gemessenen Frequenzen. Wie verlässlich sind die berechneten Werte? Diskutieren Sie auch die berechneten Trends der IR-Intensitäten von dem Hintergrund Ihrer quantenchemischen Intuition und Ihres Kenntnis des harmonischen Oszillators. Wieso zeigen die homonuklearen Moleküle keine IR-Intensität?
2. IR- und Raman-Spektrum von Benzol:
Nun soll das Schwingungsspektrum auch für Benzol berechnet werden. Hierbei steigt die Zahl der Normalmoden für nicht-lineare Moleküle mit \(3N-6\). Dadurch werden sowohl die IR- und Raman-Spektren komplexer, als auch die numerische Berechnung der Frequenzen teurer. Dennoch können numerische Frequenzen und Polarisierbarkeiten für kleine Moleküle wie Benzol noch in kurzer Zeit berechnet werden.
Optimieren Sie die Geometrie von Benzol. Berechnen Sie anschließend die Schwingungsfrequenzen, IR- und Raman-Intensitäten. Verwenden Sie hierzu erneut die r2-SCAN-3c Methode mit den gleichen Einstellung wie in der vorherigen Aufgabe (Die Rechnung dauert ca. 30 min!).
Ordnen Sie nun die berechneten den experimentellen Frequenzen von Benzol zu. Bestimmen Die auch den Charakter der Schwingung (sym/anti-CH-Streckschwinung, CH-Deformationsschwingung oder Ringschwingung). Visualisieren Sie dazu die Schwingungsmoden mit dem Programm Molden (siehe Molden für eine kurze Einführung). Experimentelle Frequenzen:
IR |
Raman |
inaktiv |
|---|---|---|
410 |
||
605.6 |
||
673 |
||
703 |
||
848.9 |
||
975 |
||
992 |
||
1038 |
||
1150 |
||
1178 |
||
1310 |
||
1326 |
||
1486 |
||
1596 |
||
3068 |
||
3047 |
||
3063 |
||
3062 |
Hinweis
Aufgrund der Symmetrie von Benzol sind viele Schwingungsmoden entartet, sodass einigen Frequenzen mehrere Moden zugeordnet werden können (siehe den NIST-Eintrag für Benzol). Zusätzlich sind die angegebenen experimentellen Frequenzen nicht vollständig, sodass nicht alle berechneten Moden zugeordnet werden können.
Geben Sie eine durchschnittliche und maximale absolute Abweichung der berechneten Frequenzen von den experimentellen Referenzwerten an.
Plotten Sie auch das berechnet IR-Spektrum mit
orca_mapspc(siehe orca_mapspc für eine kurze Einführung). Abb. 4 zeigt ein experimentelles Referenzspektrum gemessen in der Gasphase.
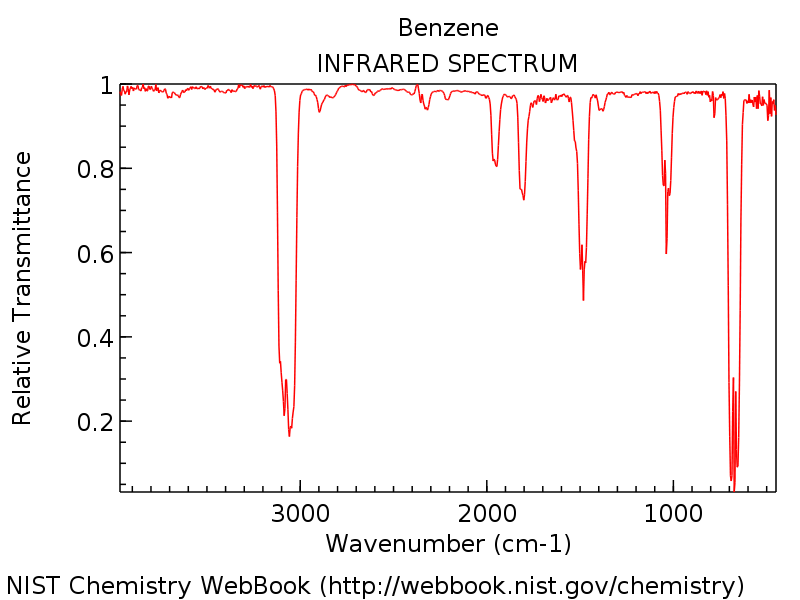
Abb. 4 Experimentelles IR-Spektrum von Benzol in der Gasphase.#
3. Schwingungspektroskopie bei Wasserstoffbrückenbindungen:
Abschließend soll nun auch eine chemische Fragestellung mit der theoretischen Schwingungsspektroskopie beantwortet werden. Dabei ist das Testsystem die Benzoesäure, die über zwei Wasserstoffbrückenbindungen verbrückt auch als Dimer vorliegen kann.
Berechnen Sie zunächst die Schwingungsspektren der Benzoesäure als Monomer in der Gasphase. Optimieren Sie die Struktur dafür zuerst wie in den vorherigen Aufgaben. Plotten Sie die beiden berechneten Spektren erneut mit
orca_mapspc. Input für die Frequenzrechnung:
! R2SCAN-3C VeryTightSCF DefGrid3
! AnFreq
%pal
nprocs 4
end
* xyzfile 0 1 geom.xyz
Berechnen Sie die Schwingungsspektren auch in CCl4. Nutzen Sie dafür denselben Input wie zuvor für die Optimierung, wobei Sie den Block
%cpcmeinfügen (siehe Frequenzinput), der Input für die Frequenzrechnung sieht wie folgt aus:
! R2SCAN-3C VeryTightSCF DefGrid3
! AnFreq
! SMD(CCl4)
%pal
nprocs 4
end
* xyzfile 0 1 geom.xyz
In Tab. 22 ist das experimentelle IR-Spektrum von Benzoesäure in Gasphase (links) und in einer 2%-igen CCl4 Lösung (rechts) abgebildet. [1] Vergleichen Sie diese mit den berechneten Spektren. Können Sie eine Aussage über das Vorliegen des Benzoesäure Monomers und Dimers machen? An welchen Banden machen Sie dies fest?
|
|
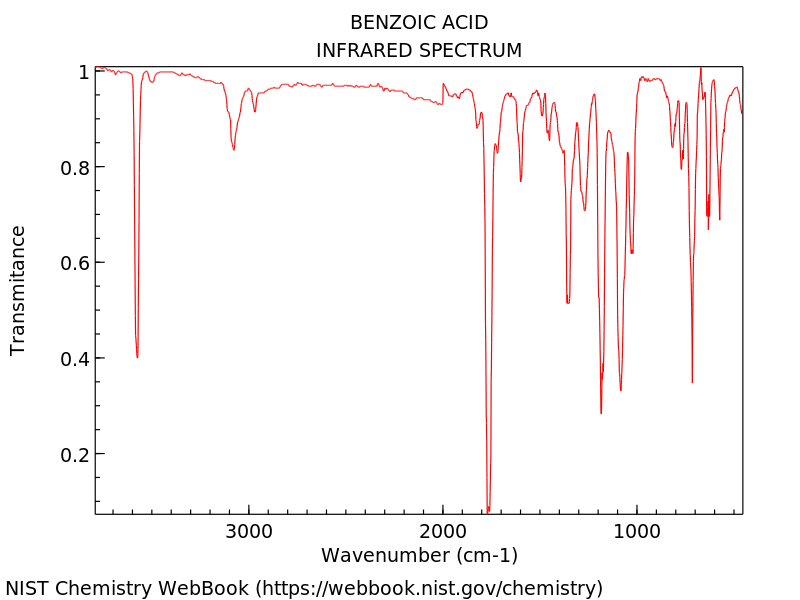
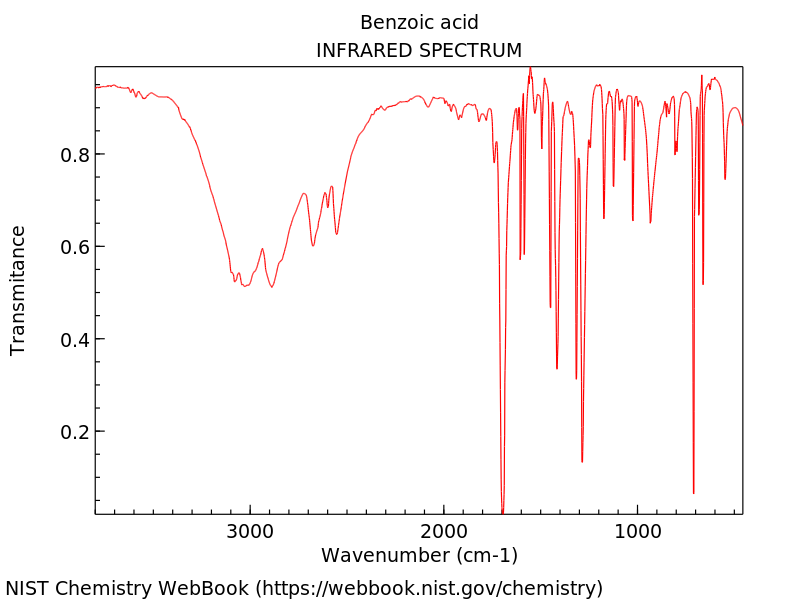
Mithilfe der sehr effizienten GFN2-xTB Methode, soll nun auch das Gasphasen IR-Spektrum des Benzoesäuredimers berechnet werden. Dafür wird auch das Dimer optimiert und dann dessen Frequenzen berechnet. Optimieren Sie dafür zunäcsht folgende Dimer Struktur mithilfe von GFN2-xTB (über das
xtbProgramm):
30
Coordinates from ORCA-job orca
O 4.08361963686171 1.42100218491426 -0.00000073226728
O 1.83133921050916 1.51376110969457 -0.00000053756247
C 2.89118961455814 -0.61275126220347 -0.00000015830084
C 1.66786596921808 -1.28923217083174 0.00000005650774
C 4.08873266661149 -1.33464374377284 -0.00000003849925
C 1.64337277394040 -2.67695964944375 0.00000040139691
C 4.05781228404398 -2.72342243751457 0.00000031385609
C 2.83755368068430 -3.39501341473763 0.00000053723225
C 2.88427383486136 0.86920719639148 -0.00000049420274
H 0.75047050342712 -0.71148827584570 -0.00000005200708
H 5.03274509066108 -0.80298251176861 -0.00000021712756
H 0.69345649532693 -3.20143513223073 0.00000056800684
H 4.98681514348201 -3.28415658889913 0.00000041400331
H 2.81696772552290 -4.48033640386284 0.00000081338830
H 4.00016402380650 2.42784908552584 -0.00000094507038
O 1.67544949026866 4.13087276718472 -0.00000067484339
O 3.92772992055675 4.03811393489232 -0.00000056319142
C 2.86787941980673 6.16462626264076 -0.00000012977032
C 4.09120303233605 6.84110723503027 0.00000008450553
C 1.67033633312375 6.88651868255242 -0.00000002372187
C 4.11569615382849 8.22883471528941 0.00000040502321
C 1.70125664287554 8.27529737658171 0.00000030086711
C 2.92151520995280 8.94688841795884 0.00000051914385
C 2.87479526761332 4.68266780421806 -0.00000047595502
H 5.00859853276044 6.26336339406959 -0.00000000626668
H 0.72632393627822 6.35485740291696 -0.00000019498359
H 5.06561240572427 8.75331024546315 0.00000056989514
H 0.77225375376621 8.83603147964344 0.00000038541034
H 2.94210110784763 10.03221140797559 0.00000077470791
H 1.75890513974578 3.12402588816740 -0.00000090017462
In der Kommandozeile müssen sie dafür folgendes eingeben (in struc.xyz sollte die obere Struktur gespeichert sein):
xtb struc.xyz --opt vtight > xtb.out
Die optimierte Struktur befindet sich nun in xtbopt.xyz. (Mit molden xtbopt.log können sie wie gehabt die Optimierung visualisieren)
Berechnen Sie nun die IR-Frequenzen mit GFN2-xTB + PTB, indem Sie die optimierte Geometrie in einen neuen Ordner kopieren und folgendes eingeben:
xtb xtbopt.xyz --ptb --hess > xtb.out
Die IR-Frequenzen befinden sich in der Datei vibspectrum.
Abschließend nutzen Sie folgenden Befehl um die IR-aktiven Moden aus dem
vibspectrumin eine Daten Datei zu schreiben:
nsm vibspectrum --plot --range 0.0 4000.0
Plotten Sie nun die erstelle plot.dat Datei mithilfe eines geeigneten Programms (Gnuplot, Excel, Libreofficecalc, …)
Wie unterscheiden sich die berechneten IR-Spektren des Monomers und des Dimers in Gasphase?
Hinweis
Im Dimer treten aufgrund der zwei enthaltenen Monomere die Moden zweifach auf. Diese können aber auch Kombinationsmoden sein und müssen nicht entartet sein.
Wasserstoffbrückenbindungen sind ein wichtiges Phänomen, beispielsweise für die Lösungsmitteleigenschaften von Wasser und das Benzoesäuredimer zeigt deren substantiellen Einfluss auf das IR-Spektrum. Das wirft für die Modellierung von IR-Spektren in protischen Lösungsmitteln wie Wasser die Frage auf, ob die häufig verwendeten impliziten Lösungsmittelmodel wie CPCM ausreichen. Dazu soll im Folgenden der Effekt von weiteren umgebenden Benzoesäuremolekülen auf die Schwingungsfrequenzen implizit modelliert werden.
Modellieren Sie also nun ein Benzoesäuremonomer in der Umgebung von Benzoesäure mit dem impliziten Lösungsmittelmodel CPCM. Setzen Sie dafür die Dielektrizitätskonstante \(\varepsilon_0=8.94\) der Wert von Benzoesäure. [2] Nutzen Sie dafür wieder ORCA und die Inputs aus Punkt 2 dieser Aufgabe., wobei Sie den
%cpcmBlock in der Inputdatei für die Optimierung und Frequenzrechnung wie folgt anpassen:
1%cpcm
2epsilon 8.94
3end
Beeinflusst das Kontinuumsmodell die Schwingungsfrequenzen der Benzoesäure in gleichem Maße wie im Benzoesäuredimer? Achten Sie dabei besonders auf die OH-Streckschwingung. Was bedeutet dies für die Anwendung von impliziten Lösungsmittelmodellen bei protischen Lösungsmitteln wie beispielsweise Wasser?